Protein Structure Prediction
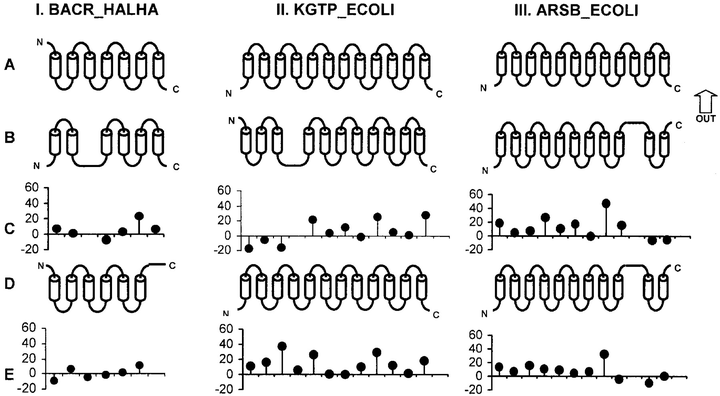 Example of predicted transmembrane segment topologies for three proteins.
Example of predicted transmembrane segment topologies for three proteins.Active from 1997 to 1999
Research rationale
Membrane proteins are involved in a wide range of essential functions, including the communication between cells and the transport of nutrients, ions, and waste products across biological membranes. These proteins, that are estimated to constitute 25% of proteins at a genomic scale, play key roles in an equally wide range of diseases like diabete, hypertension, depression, arthritis and cancer. They are also common drug targets (for over 75% of pharmaceuticals in use today). Determining membrane protein structures is essential for the understanding of how drugs interfere with cellular communication and regulation. However, current knowledge about the detailed 3D structures of membrane proteins is limited, because such protein structures are difficult to study by traditional experimental methods.
The idea is to use computational techniques to enhance our knowledge about membrane proteins. However, developing algorithms that are capable of predicting the three-dimensional structure of proteins at atomic detail is a very difficult task. Instead of tertiary structure determination, we focused our research on two complementary aspects: the structural classification of proteins, which allows to identify potential membrane proteins, and the prediction of transmembrane alpha-helices in membrane proteins.
Results
Structural classification of proteins
Periodical patterns and tandem repeats of residues are often found in DNA and protein sequences. In proteins, their presence helps towards an understanding of the molecular structure of a fibrous/structural protein employing the principle of conformational equivalence and it may suggest ways of ultramolecular assembly for the formation of higher order structure. Characteristic examples are periodicities found in a number of sequences of fibrous proteins (e.g. tropomyosin, myosin, keratins and collagen). We used the Fourier analysis method to highlight hidden periodicities in protein sequences and developped FT, a tool accessible by biologists through the Internet1 2.
In the continuation of this work, we explored the use of hierarchical, artificial neural networks for the generalized classification of proteins into several distinct classes - transmembrane, fibrous, globular, and mixed - from information solely encoded in their amino acid sequences 3 4 5. The use of our implementations (PRED-TMR2 and PRED-CLASS) to analyze various test sets and complete proteomes of several organisms demonstrates that such methods could serve as a valuable tool in the annotation of genomic open reading frames with no functional assignment or as a preliminary step in fold recognition and ab initio structure prediction methods 6 7 8 9 10 11.
Prediction of transmembrane alpha-helices in membrane proteins
The successful location of transmembrane segments, of their secondary structure and the packing modes of secondary structure elements is important because they define the architecture of a transmembrane protein. However, equally important is the determination of topology, which defines the polarity of integral membrane proteins.
Researchers have identified several characteristics that are common to a large proportion of transmembrane segments. They observed, for example, that transmembrane segments are mainly composed of hydrophobic residues, and that the propensity of positively charged residues is higher in the non-transmembrane segments on the inner part of the cell, also that a high propensity of tyrosine and tryptophan indicates the outer part of the cell. To enhance this knowledge, we performed several statistical analysis of known transmembrane segments to find other characteristics of transmembrane parts. We determined, among other things, the distribution of transmembrane segment length, the propensity for each amino acid to be in a transmembrane region and the precise profiles of potential termini (“edges”, starts and ends) of transmembrane regions [@Pasquier1999a]. We combined this information with several scoring functions to predict the precise position of transmembrane segments and their topology 12 13. The accuracy of our method compares well with that of other popular existing methods. This work led to the implementation of several tools freely available on the Internet: PRED-TMR, OrienTM, CoPreTHi and Dam-Bio.
Funding
| Program | Training and Mobility of Researchers (TMR) |
|---|---|
| Year | 1997-1999 |
| Funder | European Economic Community (EEC) |
| Grant name | EEC-TMR “GENEQUIZ”, Integrated Software System for Molecular Biologists |
| Grant id | ERBFMRXCT960019 |
| Project coordinator | Chris Sanders |
Softwares
- COPRETHI: Ensemble learning to predict transmembrane segments in proteins
- DAM-BIO: Integrated environment designed to support protein sequence and structure analysis on the Web
- DB-NTMR: Database of non transmembrane regions automatically extracted from the SwissProt database
- DB-TMR: Database of transmembrane regions automatically extracted from the SwissProt database
- FT: Analysis of periodic patterns in amino acid or DNA sequences by Fourrier transform
- ORIENTM: Topology prediction of transmembrane proteins and segments
- PRED-CLASS: System of cascading neural networks that classifies any protein into one of four possible classes: membrane, globular, fibrous, mixed
- PRED-TMR: Prediction of transmembrane domains in proteins
- PRED-TMR2: Identification of transmembrane proteins and prediction of their transmembranle domains
Related publications
- (1998). A Web Server to Locate Periodicities in a Sequence. Bioinformatics, Vol. 14, No. 8, pp. 749–50.↩︎
- (1998). A Web Interface for FT: A Tool Dedicated to the Study of Periodicities in Sequences. 20th Conference of the Hellenic Society for Biological Science.↩︎
- (1999). PRED-TMR2: An Hierarchical Neural Network to Classify Proteins as Transmembrane and a Novel Method to Predict Transmembrane Segments. 21st Conference of the Hellenic Society for Biological Sciences.↩︎
- (2001). PRED-CLASS: Cascading Neural Networks for Generalized Protein Classification and Genome-Wide Applications.. Proteins: Structure, Function, and Bioinformatics, Vol. 44, No. 3, pp. 361–9.↩︎
- (2001). PRED-CLASS: Bioinformatics Software for Generalized Protein Classification and Genome-Wide Applications. 23rd Conference of the Hellenic Society for Biological Sciences.↩︎
- (1998). CoPreTHi: A Program to Combine the Results of Transmembrane Protein Segment Prediction Methods. 20th Conference of the Hellenic Society for Biological Sciences.↩︎
- (1999). CoPreTHi: A Web Tool Which Combines Transmembrane Protein Segment Prediction Methods.. In silico biology, Vol. 1, No. 3, pp. 159–62.↩︎
- (1999). An Hierarchical Artificial Neural Network System for the Classification of Transmembrane Proteins. Protein Engineering, Vol. 12, No. 8, pp. 631–634.↩︎
- (2000). A Workbench for Computational Analysis of Protein Sequence and Structure on the Internet. 22nd Conference of the Hellenic Society for Biological Sciences.↩︎
- (2001). DAM-BIO: Bioinformatics Internet Workbench for Protein Analysis. New Modules and Applications to Biological Problems. 23rd Conference of the Hellenic Society for Biological Sciences.↩︎
- (2003). Evaluation of Annotation Strategies Using an Entire Genome Sequence. Bioinformatics (Oxford, England), Vol. 19, No. 6, pp. 717–26.↩︎
- (1999). OrienTM: A Novel Method to Predict Transmembrane Protein Topology. 21st Conference of the Hellenic Society for Biological Sciences.↩︎
- (2001). A Novel Tool for the Prediction of Transmembrane Protein Topology Based on a Statistical Analysis of the SwissProt Database: The OrienTM Algorithm. Protein Engineering, Vol. 14, No. 6, pp. 387–390.↩︎
Claude Pasquier
Researcher in Computer Science / Computational Biology
Université côte d’Azur, CNRS, I3S Laboratory, Sophia Antipolis