Omics Data Mining
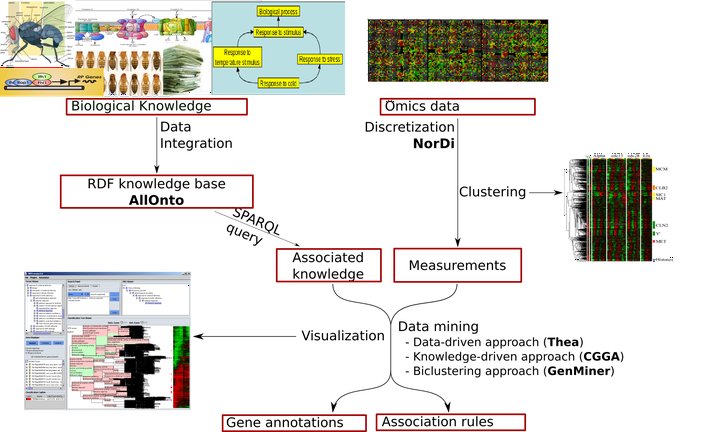 Overview of our analysis pipeline.
Overview of our analysis pipeline.Active since 2000
Research rationale
In the late 1990s, new techniques for measuring the expression of genes at the level of entire genomes have emerged. By combining these quantitative measurements with biological knowledge, this breakthrough has paved the way for deciphering the activity of genes, their interactions and their involvement in various biological processes. However, the analysis of this mass of data remained a manual task. Firstly, because, although there were many different sources storing biological data, all these sources were completely independent of each other, and secondly, because tools to analyse these data in an automated way did not yet exist. Solutions to integrate heterogeneous data and to automate their analysis were more than ever needed.
Results
A methodology to ease data integration using Semantic Web technologies
Our research started with the idea that Semantic Web technologies, which provide a common framework allowing data to be shared and reused between applications, might be applied to the management of disseminated biological data. We studied and reported the specificities of biological data that made the application of these technologies to the life sciences a real challenge. Then, we proposed a methodology to facilitate data integration using Semantic Web whose precepts were very close to the rules that currently govern the Web of Data 1 2. We implemented our ideas in AllOnto, a Knowledge Base System capable of storing and performing queries on large sets of RDF/OWL specifications (including the storing and querying of reified statements). The software was designed to handle the provenance of information and included reasoning capabilities dealing with type inference, transitivity and built-in OWL constructs like owl:sameAs and owl:inverseOf.
Allonto was applied to collect and integrate data used in three different approaches of data mining we investigated.
Automatic gene annotation through a data-driven approach
The data-driven approach involves first identifying groups of genes whose expression shows similar variation and then integrating knowledge about genes. The research on this topic took the form of an integrated system called THEA (Tools for High-throughput Experiments Analysis). The software integrates several data mining algorithms to automatically annotate groups of genes sharing similar expression profiles with biological information (various ontologies, chromosomal localization, link with diseases). Experiments show that using THEA not only makes it easy and quick to obtain all manually highlighted results, but also to pinpoint new findings 3 4.
Automatic gene annotation through a knowledge-driven approach
The knowledge-driven approach consists of first finding co-annotated groups of genes and then, in a second step, integrating data on expression profiles. The CGGA method (Co-expressed Gene Groups Analysis), which we developed, is part of this approach. The tests that have been carried out show that the functional annotations provided by CGGA reduce the complexity of the data analysis problem by integrating various types of information about genes. The experimental results showed the interest of the approach and made it possible to identify relevant information on the biological processes studied 5 6 7 8 9 10.
Extraction of association rules from a heterogeneous set of gene data
We have proposed the use of Association Rule Discovery (ARD) as a method capable of identifying rules linking any pieces of biological data and which does not impose any ordering over the use of data sources. We developed an application called GenMiner to fully exploit the capacities of ARD in the context of biological data mining. GenMiner allows the joint use of knownledge about genes and their level of expression under certain conditions in order to discover the relationships between a priori knowledge and experimental measurements. Our method includes a new algorithm, called NorDI (Normal DIscretization algorithm) to discretize gene expression measurements and generate expression profiles. The experiments we conducted confirmed the advantages of GenMiner over known approaches. GenMiner allows to search for association rules using a much smaller minimum support than what is possible with traditional approaches. In addition, GenMiner significantly reduces the number of extracted rules, making it much easier for the end user to explore and interpret 11 12 13.
Alongside these activities, research was also carried out on the parallelization of the Blast algorithm 14. Collaborations with biologists still continue, resulting in collaborative research where we are primarily responsible for data analysis 15 16 17 18 19 20 21.
Funding
| Program | Inter-EPST Program on bioinformatics |
|---|---|
| Year | 2002-2004 |
| Funder | CNRS, INSERM, INRA, INRIA, Ministry of Research |
| Grant name | The use of a knowledge base system to analyze microarray data |
| Project coordinator | Claude Pasquier |
| Program | CNRS Bio-STIC-LR |
|---|---|
| Year | 2005-2007 |
| Funder | CNRS, INSERM, INRA, INRIA, Ministry of Research |
| Grant name | Towards an editor for the subdivision of trees into sub-trees collections formals and functionals criteria for the subdivision process, intra-inter collection trees comparaisons |
| Project coordinator | François Chevenet |
| Program | CNRS post-doctoral grant |
|---|---|
| Year | 2008-2010 |
| Funder | CNRS |
| Grant name | Transcriptome mass data use and interpretation using the Massively Parallel Signature Sequencing (MPSS) technologies |
| Grant recipient | Ronnie Alves |
| Project coordinator | Claude Pasquier |
| Program | ANR Methylclonome |
|---|---|
| Year | 2013-2015 |
| Funder | ANR |
| Grant name | Analyse de l’héritabilité des traces épigénétiques dans la reproduction clonale |
| Grant id | ANR-12-BSV6-0006 |
| Project coordinator | Alain Robichon |
| Program | Gliosplice |
|---|---|
| Year | 2017-2020 |
| Funder | Institut National du Cancer (INCa) |
| Grant name | Characterization of alternative splicing networks coordinating brain tumor heterogeneity and treatment resistance commitment |
| Project coordinator | Mathieu Gabut |
Softwares
- AllOnto: Knowledge Base System to store and query RDF/OWL specifications
- CGGA: Extraction of bi-clusters of genes
- GenMiner: Mining equivalence classes and minimal non-redundant association rule from gene expression data
- NORDI: Discretization of gene expression data according to the distribution of the dataset
- THEA: Integrated information processing system dedicated to the annotation of transcriptomic results
- Thea-Interact: Analysis of the interaction network of Drosophila genes
- Thea-Online: Web portal using Semantic Web technologies to integrate, query and display information from multiple sources
Related publications
- (2008). Biological Data Integration Using Semantic Web Technologies.. Biochimie, Vol. 90, No. 4, pp. 584–94.↩︎
- (2011). Applying Semantic Web Technologies to Biological Data Integration and Visualization. Data Management in Semantic Web, pp. 131–151.↩︎
- (2004). THEA: Ontology-Driven Analysis of Microarray Data.. Bioinformatics (Oxford, England), Vol. 20, No. 16, pp. 2636–43.↩︎
- (2005). Exploratory Analysis of Cancer SAGE Data. 9th European Conferences on Principles and Practice of Knowledge Discovery in Databases (PKDD'05), Discovery Challenge.↩︎
- (2006). Co-Expressed Gene Groups Analysis (CGGA): An Automatic Tool for the Interpretation of Microarray Experiments. Journal of Integrative Bioinformatics, Vol. 3, No. 2, pp. 1–12.↩︎
- (2006). Analyse des groupes de gènes co-exprimés (AGGC): un outil automatique pour l'interprétation des expériences de biopuces. 13ème Rencontres de la Société Francophone de Classification (SFC’06), pp. 267–276.↩︎
- (2008). Analyse des groupes de gènes co-exprimés: un outil automatique pour l'interprétation des expériences de biopuces (version étendue). Revue des Nouvelles Technologies de l’Information (RNTI-C-2), Classification : points de vue croisés, Vol. 831, pp. 263–74.↩︎
- (2008). Mining Gene Expression Data Using Domain Knowledge. International Journal of Software and Informatics (IJSI), Vol. 2, No. 2, pp. 215–231.↩︎
- (2009). Mining Association Rule Bases from Integrated Genomic Data and Annotations (Extended Version). Lecture Notes in Bioinformatics, Vol. 5488, pp. 78–90.↩︎
- (2007). GenMiner: Mining Informative Association Rules from Genomic Data. IEEE International Conference on Bioinformatics and Biomedicine (BIBM'07), pp. 15–22.↩︎
- (2008). GenMiner: Mining Non-Redundant Association Rules from Integrated Gene Expression Data and Annotations.. Bioinformatics (Oxford, England), Vol. 24, No. 22, pp. 2643–4.↩︎
- (2008). Mining Association Rule Bases from Integrated Genomic Data and Annotations. 5th International Conference on Computational Intelligence Methods for Bioinformatics and Biostatistics (CIBB'08), pp. 33–43.↩︎
- (2014). Environmentally Selected Aphid Variants in Clonality Context Display Differential Patterns of Methylation in the Genome. PLOS ONE, Vol. 9, No. 12, pp. e115022.↩︎
- (2020). Computational Prediction of miRNA / mRNA Duplexomes at the Whole Human Genome Scale Reveals Functional Subnetworks of Interacting Genes with Embedded miRNA Annealing Motifs. Computational Biology and Chemistry, Vol. 88, pp. 107366.↩︎
- (2022). L’essentiel du symposium UCAncer 2021 sur les approches multidisciplinaires de la recherche sur le cancer. Bulletin du Cancer, Vol. 109, No. 4, pp. 505–509.↩︎
- (2022). Persistent Properties of a Subpopulation of Cancer Cells Overexpressing the Hedgehog Receptor Patched. Pharmaceutics, Vol. 14, No. 5, pp. 988.↩︎
- (2022). SETMAR Shorter Isoform: A New Prognostic Factor in Glioblastoma. Frontiers in Oncology, Vol. 11, pp. 638397.↩︎
- (2023). CELF2 Sustains a Proliferating/OLIG2+ Glioblastoma Cell Phenotype via the Epigenetic Repression of SOX3. Cancers, Vol. 15, No. 20, pp. 5038.↩︎
- (2023). A Machine Learning Approach to Predict Weaning Outcome among Ventilated Patients in Intensive Care Unit. Reanimation 2023, the French Intensive Care Society International Congress, Vol. 13.↩︎
Claude Pasquier
Researcher in Computer Science / Computational Biology
Université côte d’Azur, CNRS, I3S Laboratory, Sophia Antipolis